

Illustrations by Eva Giurco
What is it?
Desmoid fibromatosis (DF), also called desmoid tumor or aggressive fibromatosis, is a rare form of tumor, resulting from tendons and musculo-aponeurotic structures. FD results from the clonal proliferation of fibroblasts, connective tissue cells that support tissue and play a role in wound healing. Mutations in the DNA of these cells cause their uncontrolled growth leading to the formation of desmoid tumors. It’s a tumor characterized by infiltrative growth and a tendency towards local recurrence, that’s why it’s also referred to as “aggressive fibromatosis” but doesn’t have the potential for metastasis, that is the ability to spread to other tissues. However, sometimes DF can be multifocal.
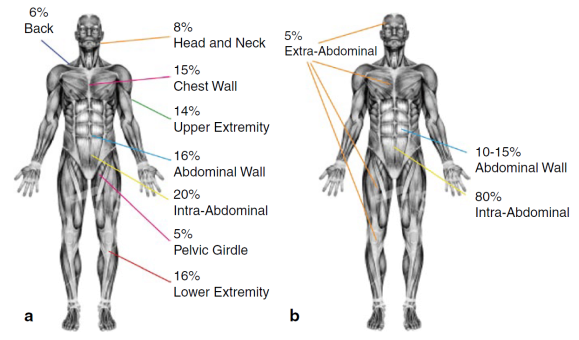
Desmoid fibromatosis can occur anywhere in the body and more than one tumor can grow up in the same site. The localization of the disease can be:
EXTRA-ABDOMINAL:neck, shoulders, limbs, gluteal region, are the sites with highest percentage of recurrence after surgical excision.
ABDOMINAL WALL: originates from the muscle bands or from the abdominal/thoracic wall
INTRA-ABDOMINAL: in mesentery or retroperitoneum, it’s the rarer site where the tumor can grow up and, growing deep in the abdomen, it can lead to compression and/or obstruction of internal organs.
Desmoid tumor Distribution
Desmoid fibromatosis (DF) is a rare tumor: its annual incidence is 2-4 cases/1 million in the population and represents 0.03% of all neoplasms. This explains the difficulty in diagnosing for many doctors and above all the importance of being followed by a dedicated specialist, who knows the disease and that can follow the patient with the best diagnostic/therapeutic approach. DF can affect persons of all ages, but is more common in adults between 18 and 35 years and, in particular, in women of childbearing age, often with a recent history of childbirth or surgery.
Sono stati chiamati in causa, infatti, vari fattori eziopatogenetici, tra cui: traumi, anche di tipo chirurgico, in soggetti con squilibri endocrini e/o predisposizione genetica alle connettivopatie.
Several etiopathogenetic factors seem likely to be responsible for this tumor, including: trauma, also considering surgical ones, in persons with endocrine imbalances and/or genetic predisposition to connective tissue diseases.
In most cases, desmoid tumor has a SPORADIC origin, but there is a small percentage of persons (5-10%) with DF who have a familial syndrome called Familial Adenomatous Polyposis (FAP) or Gardner’s Syndrome. Patients affected by this syndrome usually discover that they have DF during routine screening tests.
We distinguish:
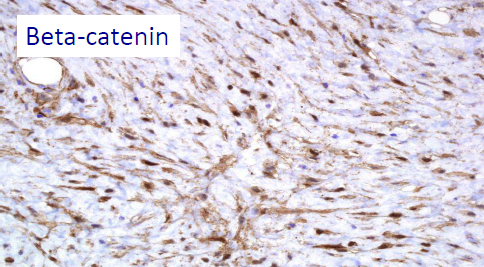
– SPORADIC CASES OF DESMOID TUMOR: the cause is not known. In 85% of cases, somatic mutations were identified in the CTNNB1 gene (3q21), a gene that codes for β-catenin (i.e. mutations occurred in a single cell that is transmitted to his cells proliferative line, but the mutation is NOT transmitted from the patient to his children). In this case, there is the presence of the desmoid tumor without other clinical manifestations, as it happens in FAP.
– DESMOID TUMORS IN PATIENTS WITH FAP: desmoid tumors that appear during FAP (familial adenomatous polyposis), represent only 10% of all cases of desmoid tumors. In this case, the mutation is localized in a germ cell and it concerns the tumor suppressor gene APC, responsible for the control of cell growth and death, present on chromosome 5 (5q21-q22), which encodes the adenomatous polyposis polyposis protein of the colon, a protein therefore mutated into all the patient’s cells. For this reason, the syndrome can be transmitted from the patient to his children.
The β-catenin and APC mutations seem to EXCLUDE each other, so the identification of a somatic mutation of the β-catenin can help to exclude a systemic disease such as FAP. Thus, the patient will have either a sporadic desmoid mutation – that is, the β-catenin mutation – or the APC mutation typical of FAP; he cannot have both of them. Instead, the wild type state (absence of mutation) of β-catenin should arouse suspicion of FAP; in this case it’s recommended to study familial history for FAP and/or to do a colonoscopy.
CTNNB1 mutation of beta-catenin
Present in 85% of sporadic desmoid tumors
Molecular level:
Both CTNNB1 and APC act on the WNT pathway, a molecule that promotes the degradation of β-catenin.
- In the normal WNT pathwayt: when Wnt ligand arrives on the extracytoplasmic side of its membrane receptor, a protein called Disheveled (Dvl) binds to the receptor on the intracellular surface. A multi-protein complex is formed including APC, glycogen synthase kinase (GSK-3β) and Axin which bind β-catenin and this one is then phosphorylated by GSK-3β and degraded by the proteasome.
- However, if there is a mutation on CTNNB1 or APC, the multi-protein complex cannot be generated, leading to the stabilization of the β-catenin protein which is neither phosphorylated nor degraded. Consequently, β-catenin accumulates in the cytoplasm of the cell and afterwards it translocates to the nucleus, where it binds to transcription factors involved in cell proliferation takes place, and also cyclin D1 and COX-2 are activated, with a proliferative and anti-apoptotic role.
DF

WARNING! Informations on this site are designed to improve and to inform the patient and in general to promote greater awareness of their pathology. In no case they replace specialist medical advice. While guaranteeing the accuracy and scientific rigor of the information, DESMOID FOUNDATION declines all responsibility about informations provided on the treatments, reminding all patients to contact their specialist doctor and to evaluate with him the best treatment in a specific personal case.