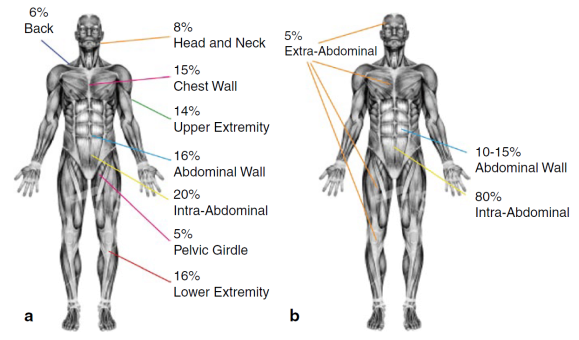
La FD è un tumore raro, la sua incidenza annuale varia da 2-4 casi per milione nella popolazione e rappresenta lo 0,03% di tutte le neoplasie. Ciò spiega la difficoltà di molti medici nella diagnosi e soprattutto l’importanza di essere seguiti da uno specialista dedicato, che conosca la patologia ed il miglior approccio diagnostico/terapeutico.La FD può interessare qualsiasi fascia di età, ma è più comune negli adulti tra i 18 e 35 anni e, in particolar modo, nelle donne in età fertile, che spesso hanno in anamnesi recente parto o intervento chirurgico.
Sono stati chiamati in causa, infatti, vari fattori eziopatogenetici, tra cui: traumi, anche di tipo chirurgico, in soggetti con squilibri endocrini e/o predisposizione genetica alle connettivopatie.
Nella maggior parte dei casi, il tumore desmoide ha un’origine SPORADICA, ma c’è una piccola percentuale di persone (il 5-10%) affette da FD, che hanno una sindrome familiare chiamata Poliposi adenomatosa familiare (FAP) o sindrome di Gardner. I pazienti affetti da queste sindromi ero-familiari usualmente scoprono di avere la FD in corso di esami di routine eseguiti per screening.
Distinguiamo:
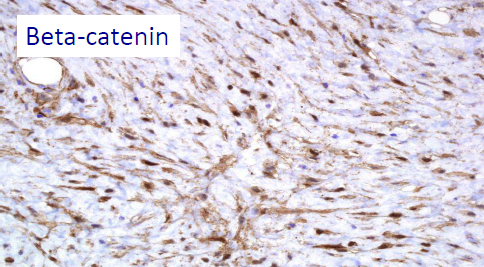
– I CASI SPORADICI DI TUMORE DESMOIDE: vengono definiti così poiché la causa non è conosciuta. Nel 85% dei casi sono state individuate mutazioni somatiche del gene CTNNB1 (3q21), gene che codifica per la beta-catenina (cioè mutazioni avvenute in una singola cellula che la trasmette alle sue cellule figlie, ma la mutazione NON viene ereditata dalla prole del paziente). In questo caso c’è la presenza del solo tumore desmoide senza contemporanea presenza di altre manifestazioni cliniche, come invece avviene nella FAP.
– TUMORI DESMOIDI IN PAZIENTI PORTATORI DI FAP: I tumori desmoidi che insorgono nel contesto della FAP (poliposi adenomatosa familiare), rappresentano solo il 10% di tutti i casi di tumore desmoide. In questo caso la mutazione è a livello germinale e riguarda il gene oncosoppressore APC, deputato al controllo della crescita e morte cellulare, presente sul cromosoma 5 (5q21-q22), che codifica per la proteina della poliposi adenomatosa del colon, proteina quindi mutata in tutte le cellule del paziente. Per questo motivo, la Sindrome può essere trasmessa.
Le mutazioni beta catenina e APC sembrano ESCLUDERSI a vicenda, quindi l’identificazione di una mutazione somatica della beta catenina può aiutare ad escludere che nel paziente ci sia una condizione sistemica come la FAP. Perciò il paziente avrà o una mutazione per il desmoide sporadico – cioè la mutazione della beta catenina – o la mutazione APC tipica della FAP; non potrà averle entrambe. Viceversa lo stato wild type (assenza di mutazione) di beta catenina, dovrebbe destare sospetti FAP; in questo caso è consigliato studiare anamnesi familiare per FAP e/o colonscopia per escluderla.